Excited State Proton Transfer in 2’-hydroxchalcone derivatives
by Michael Dommett, 4/01/2017
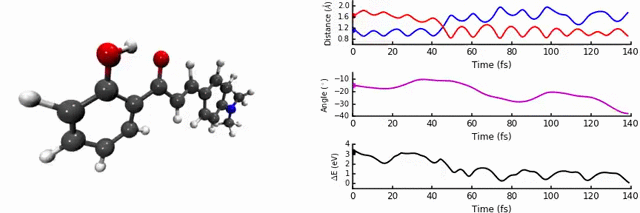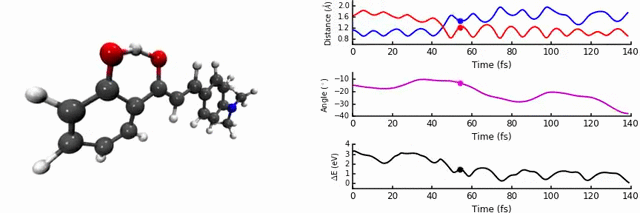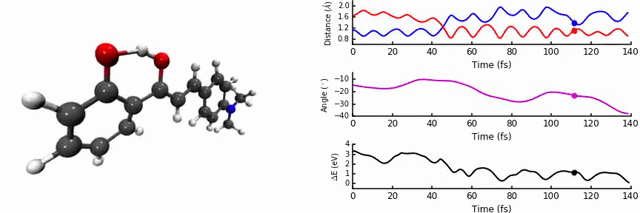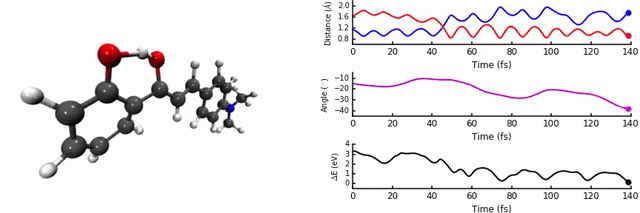

In our group, we are interested in modelling the photochemistry of molecules which undergo some kind of ‘switching’ process when they absorb light, for use in photochromic materials. In October 2015, we were intrigued by photochromes based on 2’-hydroxychalones. When exposed to UV-light, these systems undergo excited state intramolecular proton transfer (ESIPT), a highly interesting and useful phenomenon which can be used in optoelectronics, imaging, and solid state lasers.
The experimental data suggest that the molecular structure (electron donating substituents) and the way the molecules aggregate in the solid state determine whether the crystal fluoresces or not. In a good solvent, the molecules do not fluoresce at all. Little theoretical data exists for these compounds, and we decided to undertake a study to understand the photochemistry of these systems. In particular, we wanted to understand why some 2’-hydroxychalcones fluoresce, and others do not. For that, we needed to decouple the effect of the molecular substituent and the effect of the packing mode.
The first step of the study is to understand the photochemistry 2’-hydroxychalcones and the influence of different groups (methyl and methoxy) on the potential energy surface. This is the work we present in the paper. We studied 5 compounds in gas phase, focussing on the important steps in the photochemical process: UV-absorption, proton transfer, and relaxation. We also used non-adiabatic dynamics to understand the rate of proton transfer and the examine the relaxation pathways after absorption.
As discussed in the paper, we uncover two relaxation channels in the first excited state (S1) which cause the S0 and S1 states to converge. The first pathway involves the expected proton transfer process followed by intramolecular rotation. The conical intersection (point of degeneracy between S0 and S1) is easily accessible, resulting in non-radiative relaxation to S0 (i.e no fluorescence). The second pathway results in a conical intersection being reached via intramolecular rotational but without the proton transfer step.
The identity and position of the substituent determines which of these modes is preferred. We find that the para position on the ring is sensitive to a strong electron donating group, as the conjugation changes the character of the S1 state and favours the proton transfer path, a conclusion reached thanks to the non-adiabatic dynamics. We carefully analysed the potential energy surface with different levels of theory, in particular the region of the conical intersections. This is one of the most important aspects of the paper, as the easy-to-access conical intersections are the key in determining the fluorescence.
Whenever I read a paper in the literature, it appears that all of the pieces fell beautifully in to place for the authors and the final product reflects exactly how the work was carried out. I have now learned first-hand that this is (almost never) the case. In January 2015, I started writing the paper thinking that I already had most of the data. In fact, it took another six months of calculations to finally complete the picture. The most time-consuming (and frustrating) part was understanding the effect of method and basis set, as nothing seemed to match in the beginning. It was only through really understanding the strengths and limitations of each method did the data make sense. The characterisation of the conical intersection geometries was also quite problematic. I located at least 40 conical intersections (5 compounds, 2 methods, 2 basis sets, 2 related structures dependent on +/- rotation angle), with additional points calculated based on the dynamics data. This was really tricky and quite labour intensive.
The most enjoyable part was the running and analysis of the dynamics calculations. I used Jupyter notebooks with python/matplotlib to interpret the data, which allowed me to write scripts to analyse and plot on-the-fly. It was a workflow which was really efficient, as a slight modification to the analysis codes could easily produce new figures, something I was thankful for upon returning to the data ~4 months later to respond to the reviewers’ questions.
Going forward, we are almost ready to start writing the paper for the second step of this study. We have lots of data which I have been collecting and analysing over the last 5 months or so. It should produce a really good paper once the final holes are filled in. Watch this space.
