
Category Archives: Recent papers

Switching the Spin State of Pentafluoro phenylnitrene: Isolation of a Singlet Arylnitrene Complex

J. Am. Chem. Soc., 140, 49, 17271-17277.
The chemistry of arylnitrenes is dominated by their triplet ground states and excited open-shell singlet states. This results in radical-type reactions and unwanted rearrangements which diminish the use of arylnitrenes as intermediates in organic synthesis. While the closed-shell singlet states of arylnitrenes are expected to undergo useful chemical transformations (comparable to the closed shell singlet states of carbenes), these states are too high in energy to be chemically accessible. When triplet pentafluorophenylnitrene is interacting with the Lewis acid BF3 under the conditions of matrix isolation, a Lewis acid-base complex consisting of the closed-shell singlet state of the nitrene and two molecules of BF3 is formed. Although the closed shell singlet state of pentafluorophenylnitrene is calculated (CCSD(T)) to lie more than 25
kcal/mol above its triplet ground state, the reaction with BF3 results in switching the spin state from triplet to singlet. The formation of the singlet complex was monitored by IR, UV-vis, and EPR spectroscopy. DFT, CCSD(T), and CASPT2 calculations confirm the experimental findings.
Aryne-Mediated Arylation of Hantzsch Esters: Access to Highly Substituted Arylhydropyridines

Synthesis, 2018,50(23): 4591-4605
This is a full account of our studies into the generation of highly functionalised 2-aryl-1,2-dihydropyridines and 2-methylene-3-aryl-1,2,3,4-tetrahydropyridines via intermolecular aryne ene reactions of Hantzsch esters. Furthermore, exposure to excess aryne revealed unusual 3′-aryl-spiro[benzocyclobutene-1,1′-(3′,4′-dihydropyridines)]. Mechanistic insights are provided by deuterium-labelling studies and DFT calculations, whilst preliminary cytotoxicity investigations reveal that the spirocycles are selective against colon carcinomas over ovarian cancer cell lines and that all the compounds have high selectivity indices with regards to non-cancer cells.
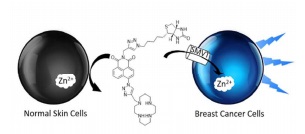
Biotin-tagged fluorescent sensor to visualize ‘mobile’ Zn2+ in cancer cells
Chem. Commun., 2018, 54, 9619-9622
A cancer cell-targeting fluorescent sensor has been developed to image mobile Zn2+ by introducing a biotin group. It shows a highly selective response to Zn2+ in vitro, no toxicity in cellulo and images ‘mobile’ Zn2+ specifically in cancer cells. We believe this probe has the potential to help improve our understanding of the role of Zn2+ in the processes of cancer initiation and development.
Recent Advances and Perspectives on Nonadiabatic Mixed Quantum–Classical Dynamics

Chem. Rev., 2018, 118, 7026–7068 (part of the Theoretical Modeling of Excited State Processes special issue).
Nonadiabatic mixed quantum–classical (NA-MQC) dynamics methods form a class of computational theoretical approaches in quantum chemistry tailored to investigate the time evolution of nonadiabatic phenomena in molecules and supramolecular assemblies. NA-MQC is characterized by a partition of the molecular system into two subsystems: one to be treated quantum mechanically (usually but not restricted to electrons) and another to be dealt with classically (nuclei). The two subsystems are connected through nonadiabatic couplings terms to enforce self-consistency. A local approximation underlies the classical subsystem, implying that direct dynamics can be simulated, without needing precomputed potential energy surfaces. The NA-MQC split allows reducing computational costs, enabling the treatment of realistic molecular systems in diverse fields. Starting from the three most well-established methods—mean-field Ehrenfest, trajectory surface hopping, and multiple spawning—this review focuses on the NA-MQC dynamics methods and programs developed in the last 10 years. It stresses the relations between approaches and their domains of application. The electronic structure methods most commonly used together with NA-MQC dynamics are reviewed as well. The accuracy and precision of NA-MQC simulations are critically discussed, and general guidelines to choose an adequate method for each application are delivered.
How Inter- and Intramolecular Processes Dictate Aggregation-Induced Emission in Crystals

J. Phys. Chem. Lett., 2017, 8, 6148–6153
Aggregation-induced emission (AIE) offers a route for the development of luminescent technologies with high quantum efficiencies. Excited-state intramolecular proton transfer (ESIPT) coupled to AIE can produce devices with emission across the visible spectrum. We use a combination of theoretical models to determine the factors that mediate fluorescence in molecular crystals undergoing ESIPT. Using two materials based on 2′-hydroxychalcone as exemplar cases, we analyze how inter- and intramolecular processes determine the emissive properties in the crystal environment. This systematic investigation extends the current interpretation of AIE to polar chromophores with multiple decay pathways. We find that population of nonradiative pathways is dictated by the electronic effects of the substituents and the degree of distortion allowed in the crystal environment. Localization of the electron density is crucial to maximize fluorescence via ESIPT. Our conclusions offer design strategies for the development of luminescent molecular crystals.
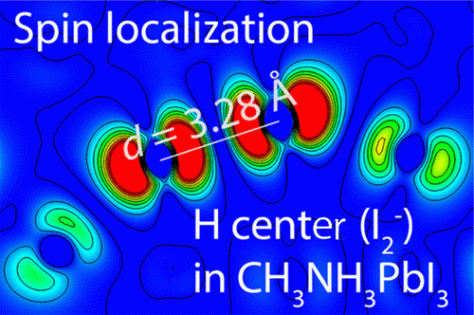
H-Center and V-Center Defects in Hybrid Halide Perovskites
ACS Energy Lett., 2017, 2, 2713–2714
The self-trapping of holes with the formation of a molecular X2– anion is a well-established process in metal halide (MX) crystals, but V-center (2X– + h+ → X2–) and H-center (X– + Xi– + h+ → X2–) defects have not yet been confirmed in halide perovskite semiconductors. The I2– split-interstitial defect is predicted to be a spin radical in CH3NH3PbI3 with an optically excited state in the semiconductor band gap.
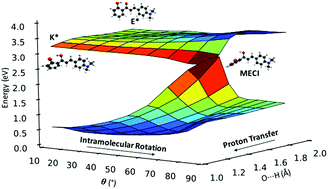
Excited state proton transfer in 2′-hydroxychalcone derivatives
Phys. Chem. Chem. Phys., 2017,19, 2409-2416
Fluorophores exhibiting excited-state intramolecular proton transfer (ESIPT) are promising candidates for applications ranging from imaging and probing to laser dyes, optoelectronic devices and molecular logic gates. Recently, ESIPT-active solid-state emitters based on 2′-hydroxychalcone have been synthesized. The compounds are almost non-emissive in solution but emit in the deep red/NIR region when crystalline. Herein, we present a comprehensive theoretical investigation of the gas-phase excited state relaxation pathways in five 2′-hydroxychalcone systems, using a combination of static and non-adiabatic simulations. We identify two competing non-radiative relaxation channels, driven by intramolecular rotation in the enol and keto excited states. Both mechanisms are accessible for the five compounds studied and their relative population depends on the nature of the substituent. The addition of electron-donating substituents greatly increases the propensity of the ESIPT pathway versus rotation in the enol state. The identification of the fundamental relaxation mechanisms is the first step towards understanding the aggregated emission phenomena of these compounds.
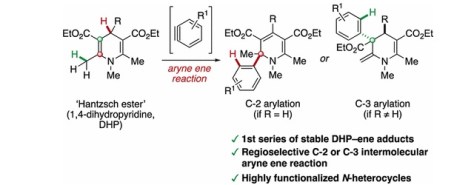
Intermolecular Aryne Ene Reaction of Hantzsch Esters
Org. Lett., 2017, 19 (17), pp 4644–4647
The reaction of arynes with 1,4-dihydropyridines affords 2-aryl-1,2-dihydropyridines or 2-methylene-3-aryl-1,2,3,4-tetrahydropyridines via a regioselective C-2 or C-3 arylation. These compounds are the first series of isolable and bench-stable covalent ene adducts formed between dihydropyridines and unsaturated substrates. Experimental studies and DFT calculations provide mechanistic support for a concerted intermolecular aryne ene process, which may have implications for NAD(P)H model reactions.
