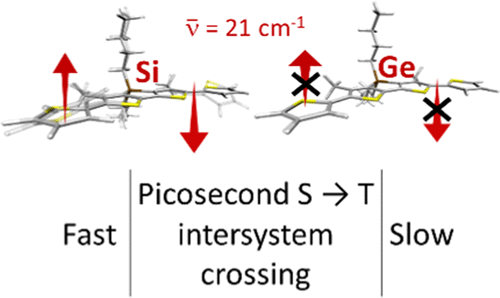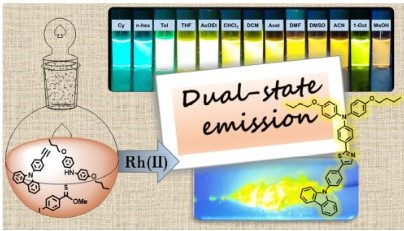
This work describes a new approach to construct highly conjugated molecules with asymmetric donor-acceptor-donor’ architectures (D-A-D’). Five new emissive compounds featuring thiazole, a scarcely used acceptor, were synthesized using a three-component Rh(II) catalytic reaction. The asymmetric fluorescent compounds show significant emission in solution (QY = 73% – 100%) and the solid-state (QY = 14% – 59%), and therefore considered Dual-State Emitters (DSE). We also evaluate the impact of O-alkyl chains with varying lengths in the photophysical properties in solution, aggregates, and solid-state. Computational studies indicate that the involved electronic transitions have a significant charge transfer character produced almost exclusively from the triphenylamine donors. According to the single crystal X-ray data of compounds 8 and 9, the conjugated structures have a twisted molecular conformation that contributes to the observed emission in the solid-state. These findings show a systematic approach to design DSE materials, which may help to stimulate their use in biological or optoelectronic applications.