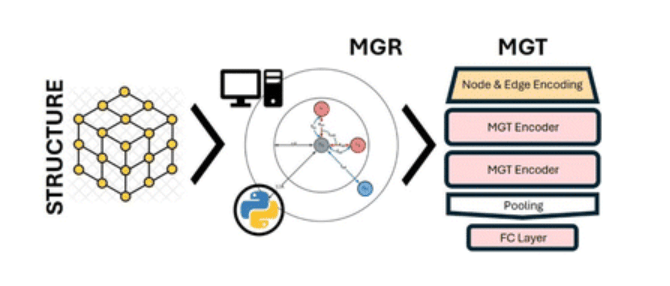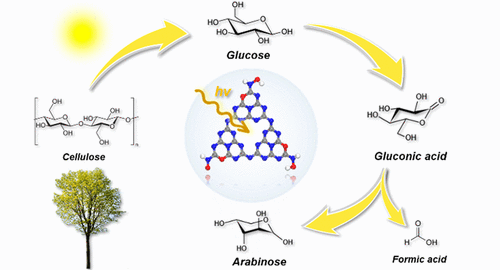
Energy Materials and Devices 2024
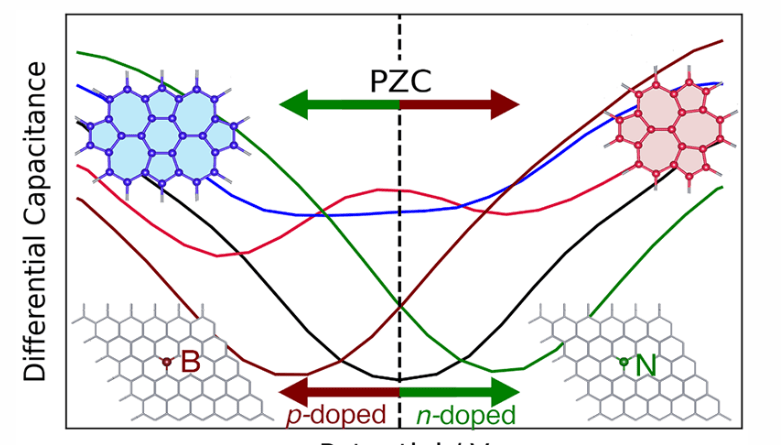
Graphene-based frameworks suffer from a low quantum capacitance due to graphene’s Dirac point at the Fermi level. This theoretical study investigated the effect structural defects, nitrogen and boron doping, and surface epoxy/hydroxy groups have on the electronic structure and capacitance of graphene. Density functional theory calculations reveal that the lowest energy configurations for nitrogen or boron substitutional doping occur when the dopant atoms are segregated. This elucidates why the magnetic transition for nitrogen doping is experimentally only observed at higher doping levels. We also highlight that the lowest energy configuration for a single vacancy defect is magnetic. Joint density functional theory calculations show that the fixed band approximation becomes increasingly inaccurate for electrolytes with lower dielectric constants. The introduction of structural defects rather than nitrogen or boron substitutional doping, or the introduction of adatoms leads to the largest increase in density of states and capacitance around graphene’s Dirac point. However, the presence of adatoms or substitutional doping leads to a larger shift of the potential of zero charge away from graphene’s Dirac point.

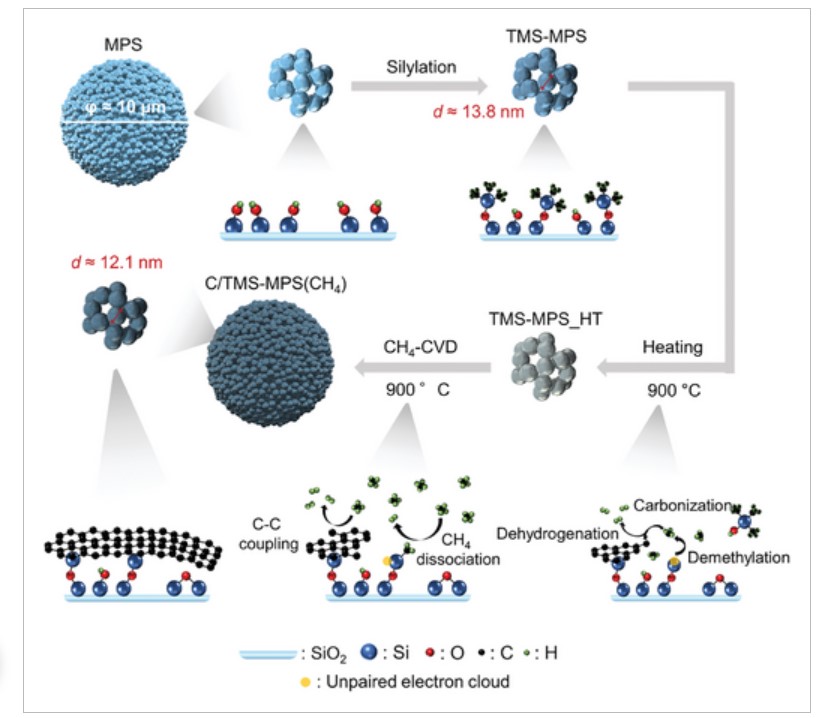
 1 by employing calcium oxide (CaO) nanoparticles as templates. The CaO nanoparticles with an approximate diameter of 86 nm are formed through the thermal decomposition of calcium carbonate nanoparticles. However, the calcium carbonate nanoparticles contain a small amount of Mg (2 wt %), and the thermal decomposition process also yields impurities, including Mg. When the CaO nanoparticles, including the Mg-based impurities, are subjected to chemical vapor deposition, the CaO surface can be coated with a thin carbon layer, primarily consisting of single-layer graphene through the specific catalysis of the CaO surface in facilitating the CH4-to-C conversion reactions. However, at the same time, the presence of Mg-based impurities leads to the formation of low-crystalline carbons, which have a detrimental effect on the subsequent high-temperature annealing at 1800 °C, following the template removal process, resulting in an excessive number of edge sits in the GMS. We have found that the harmful low-crystalline carbons can be eliminated through heat treatment in air at 350 °C. By adopting such a removal process, high-quality GMS with a minimal number of edge sites can be produced.
1 by employing calcium oxide (CaO) nanoparticles as templates. The CaO nanoparticles with an approximate diameter of 86 nm are formed through the thermal decomposition of calcium carbonate nanoparticles. However, the calcium carbonate nanoparticles contain a small amount of Mg (2 wt %), and the thermal decomposition process also yields impurities, including Mg. When the CaO nanoparticles, including the Mg-based impurities, are subjected to chemical vapor deposition, the CaO surface can be coated with a thin carbon layer, primarily consisting of single-layer graphene through the specific catalysis of the CaO surface in facilitating the CH4-to-C conversion reactions. However, at the same time, the presence of Mg-based impurities leads to the formation of low-crystalline carbons, which have a detrimental effect on the subsequent high-temperature annealing at 1800 °C, following the template removal process, resulting in an excessive number of edge sits in the GMS. We have found that the harmful low-crystalline carbons can be eliminated through heat treatment in air at 350 °C. By adopting such a removal process, high-quality GMS with a minimal number of edge sites can be produced.