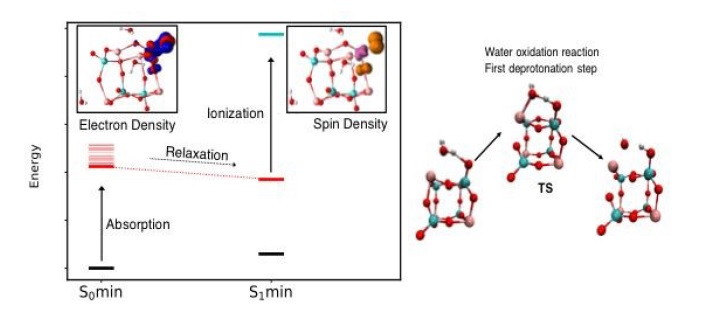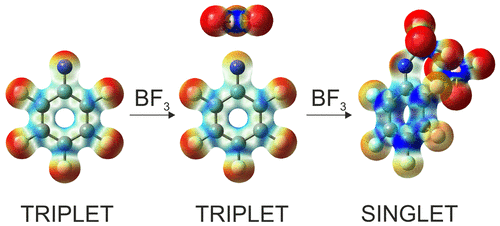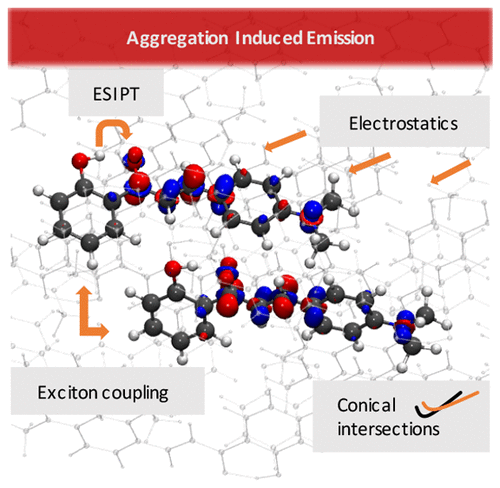
ChemPhotoChem, 2019, Just Accepted, DOI:10.1002/cptc.201900075
Organic fluorophores with an enhanced emission in the condensed phase have great potential for the design of optoelectronic materials. Several propeller-shaped molecules show aggregation-induced emission (AIE), in particular, silole derivatives have attracted significant attention because of their significant quantum yields in the solid state. In this contribution, we investigate the mechanismof AIE of a propeller-shaped blue emitter: 1,2,3,4-tetraphenyl-1,3-cyclopentadiene (TPC). We explore the excited state mechanism in the light of models most commonly used to explain it: restriction of intramolecular motions (RIM) and restricted access to the conical intersection (RACI). Our interpretation is sup-ported by excited state dynamics simulations and the analysis of Huang-Rhysfactors and reorganisation energies. We quantify the effects of intermolecular interactions and exciton couplings. The mechanism for TPCis compared with previous investigations of analogue silole compounds. Our systematic investigation highlights the role of conical intersections on the nonradiative decay mechanisms and complementary descriptions provided by the RIM and RACI models.