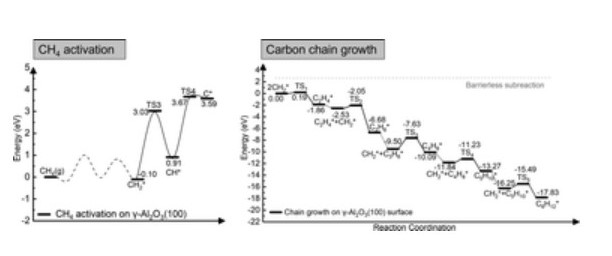
Chemical vapor deposition of methane onto a template of alumina (Al2O3) nanoparticles is a prominent synthetic strategy of graphene meso-sponge, a new class of nano porous carbon materials consisting of single-layer graphene walls. However, the elementary steps controlling the early stages of graphene growth on Al2O3 surfaces are still not well understood. In this study, density functional theory calculations provide insights into the initial stages of graphene growth. We have modelled the mechanism of CH4 dissociation on the (111), (110), (100), and (001) γ-Al2O3 surfaces. Subsequently, we have considered the reaction pathway leading to the formation of a C6 ring. The γ-Al2O3(110) and γ-Al2O3(100) are both active for CH4 dissociation, but the (100) surface has higher catalytic activity towards the carbon growth reaction. The overall mechanism involves the formation of the reactive intermediate CH2* that then can couple to form CnH2n* (n = 2–6) intermediates with unsaturated CH2 ends. The formation of these species, which are not bound to the surface-active sites, promotes the sustained carbon growth in a nearly barrierless process. Also, the short distance between terminal carbon atoms leads to strong interactions, which might lead to the high activity between unsaturated CH2* of the hydrocarbon chain. Analysis of the electron localization and geometries of the carbon chains reveals the formation of C–Al–σ bonds with the chain growing towards the vacuum rather than C–Al–π bonds covering the γ-Al2O3(100) surface. This growth behaviour prevents catalyst poisoning during the initial stage of graphene nucleation.